|
||||||
You are here: Home: Meet The Professors Vol. 4 Issue 1: Case 3

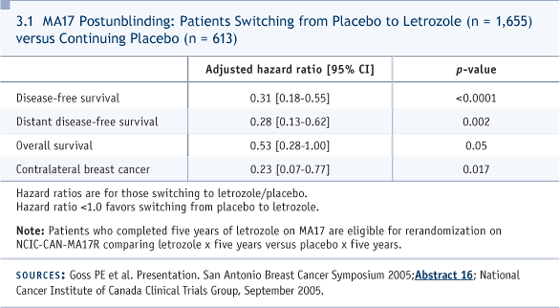
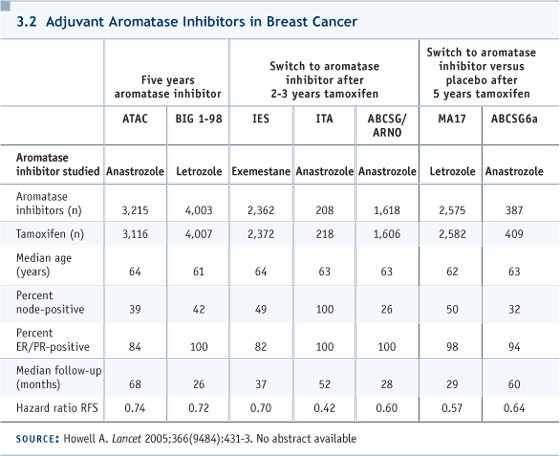
Edited excerpts from the discussion: DR ABEL: This woman was 51 in March 1988 when she was found to have a one-centimeter, ER/PR-positive infiltrating ductal carcinoma. She had undergone TAH and BSO six years previously and was receiving hormone replacement therapy and methyl testosterone at the time of diagnosis. She had implants, so a lumpectomy was not advised. She underwent a modified radical mastectomy, and her nodes were negative. She was treated with postoperative tamoxifen, since the data justifying treatment for node-negative disease had just become available. In January 2003 — 15 years later — she developed bilateral pleuritic chest pain. A chest CT showed left axillary adenopathy. A needle biopsy identified metastatic adenocarcinoma, and a bone scan was positive. Anastrozole was started in March 2003, complete resolution of pain was achieved by early June, and the CA27.29 decreased from 120 to 38 units. In early October 2004, in the context of progressively rising tumor markers, anastrozole was discontinued. The patient was treated with three doses of fulvestrant, and the markers continued to rise. Fulvestrant was discontinued, and the patient was treated with exemestane, with no benefit after three months. DR LOVE: What was her clinical condition at that point? DR ABEL: Satisfactory. She has chronic lung disease, and that’s the only comorbidity. She has no tumor-related symptoms. DR LOVE: This case is fascinating and interesting historically. This patient was in the first wave who received adjuvant endocrine therapy in 1988 for node-negative disease, and she received tamoxifen for five years. She relapsed 10 years after completing the tamoxifen. John, I’m curious about your reflections on this case, the issue of late recurrence, and what you think about the evolving data from the MA17 trial (Goss 2003, 2005b; Ingle 2005; [3.1]). DR MACKEY: This is an extraordinarily good example of why we don’t talk about breast cancer cure in the clinic, even though that’s what everyone wants to hear. They may have their five-year celebration, but we all know that these late relapses do happen. This case points out that five years of tamoxifen is probably insufficient for the ER-positive population. This woman was postmenopausal when she started her tamoxifen. If we were to see her today, of course, we would integrate an aromatase inhibitor into her treatment, and if we were seeing her after completing five years of tamoxifen, the data are there for letrozole. A small data set also exists for anastrozole in that setting (Jakesz 2005; [3.2]). We would routinely recommend letrozole. What’s particularly interesting is that at San Antonio, an analysis was presented of those women on the MA17 trial who had been randomly assigned to placebo. At the time the data came out, they were offered the option to go on open-label letrozole in an adjuvant setting, even though they’d had a treatment-free interval much longer than the three months mandated by the trial.
The analysis presented at San Antonio suggested that women who went on to letrozole — even though it was a late extended or a deferred extended letrozole treatment (Goss 2005a; [3.2]) — benefited much more than those who chose not to. In fact, a survival signal appeared when multivariate analysis was done in an attempt to correct for the baseline differences in the population. I would suggest to you that there’s no biological reason why that signal isn’t real, but it’s probable that a portion of these women have micrometastatic disease that could be suppressed by an aromatase inhibitor and would be doing better; however, I have concerns about the trial design. In the cardiology literature, people who are noncompliant with their placebos die more frequently than people who are compliant with their placebos. It’s a known fact that the kind of people who seek medical attention and follow their doctors’ orders and take recommendations live longer. It’s hard to adjust for that in multivariate analysis. So the fact that the late extended adjuvant letrozole outperformed extended letrozole suggests to me a huge bias in that analysis. So be cautious when you tell women, even if they’ve been off tamoxifen for some years, that putting them back on would give them a survival advantage. DR LOVE: I interviewed Paul Goss, who presented these data in San Antonio, and asked him whether we are moving towards a situation in which, when we see a woman like this in follow-up who’s five or 10 years past five years of tamoxifen and doing well, we should be thinking about an aromatase inhibitor? He said that we are moving in that direction. We don’t have the clinical trial evidence right now to justify doing this in practice, but we have enough data to suggest that we should be conducting research to answer that question. I know you’re hoping to approach that question using fulvestrant. Cliff, can you talk about that? DR HUDIS: These data remind us, again, of the chronic, unremitting risk of recurrence for patients with ER-positive disease. I always describe it to patients as a fixed-rate mortgage, for which the interest rate doesn’t change year after year after year. That’s the risk of recurrence. For ER-negative disease, the risk is like a bad adjustable-rate mortgage, with which you have a big balloon up front and then the risk rate drops tremendously. For these patients whom you have to imagine have microscopic metastatic disease that’s popping up clinically at a very regular rate, intervening with an active therapy, compared to not intervening, should at any point in time lower their risk of recurrence. We don’t have data for that, but we are, indirectly, gaining it. For example, it doesn’t seem to matter when you start an aromatase inhibitor compared to five years of tamoxifen in each of the three scenarios tested: up front, in the middle and out back. You nudge the hazards in a favorable direction (3.2). Similarly, MA17 is now being rerandomized for the second five years, meaning the third five years of treatment — years 11 to 15 — and obviously a hypothesis exists that more therapy at that point will continue to nudge the hazards. With all that as background, we have a hypothesis that, if we intervene with effective hormone therapy for patients who still have risk of recurrence, independent of when they finished their treatment, we should be able to demonstrate a reduction. We hope to offer fulvestrant to patients for two years beginning at some point after they finish five years of standard adjuvant therapy and demonstrate that the hazards of recurrence decrease. The hope is to prove the principle, which would allow you to then see the patient who wanders in from the outback and says, “Gee, my doc didn’t give me hormone therapy for the last four years, and should I have had it?” If we had a positive result, our answer could be a definitive “Yes.” DR LOVE: Many of the current trials of fulvestrant use loading doses. Are you planning on doing that (3.3, 3.4)? DR HUDIS: We’re still talking about it, because we’re not acutely treating established disease. An immediate or early response wouldn’t necessarily be a requirement. I hasten to add, with our focus on fulvestrant, the assumption going forward is that most of our candidate patients will have already been on aromatase inhibitors and stopped them.
DR LOVE: How do you approach the initial dosing of fulvestrant in metastatic disease? DR HUDIS: Considering the risk-to-benefit ratio for patients, we are moving in the direction of using a loading dose in the absence of randomized data proving that the loading dose is better. We give the 500-milligram dose as a first injection, and sometimes we also give a day-14 injection, and then proceed to monthly treatment. DR LOVE: I’d like to come to the next point in time. The patient has received tamoxifen, anastrozole, fulvestrant and exemestane. Correct? It sounds as though at that point you decided that she was not going to respond to additional hormones? DR ABEL: That is correct. DR LOVE: Cliff, let’s think through chemotherapy for this 66-year-old woman with asymptomatic, metastatic, hormone-resistant disease.
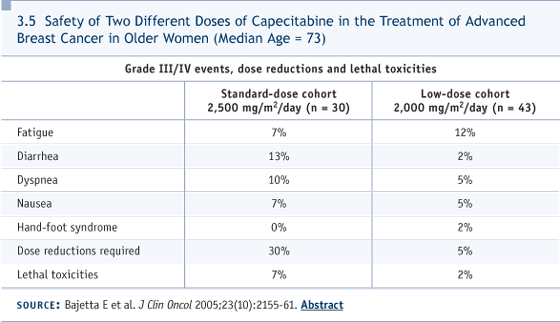
DR HUDIS: At the risk of putting a wrench in the system, I want to highlight the fact that right now we’re being driven to treatment decisions by a blood test. She has established disease, but I haven’t heard that she had progression of disease. DR ABEL: No, the imaging has not shown progression. DR HUDIS: I would not treat those blood tests. I would worry about them. I would gnaw my fingernails over them, but I wouldn’t have necessarily changed her therapy in response to them. They would have simply provoked me to keep looking harder and harder for objective evidence of progression. I don’t doubt that this patient has minimal progressive disease, but the goal of therapy for Stage IV disease remains palliation. Ideally, in this case you want to swoop in with your new therapy right before she is going to become symptomatic, whenever that is. Right now, you have no hint of objective progression. Let me push it one step further. You could practice in a community that didn’t pay for or allow you to use markers, in which case you’d be back on your first hormone therapy for her Stage IV disease, which might be just as well. DR LOVE: Unless by giving her the hormones sequentially over these years he actually prevented her from developing problems. DR HUDIS: We don’t know; it’s tough to justify treatment changes based on those blood tests. DR ABEL: Yes, this is an important issue to examine further. I don’t see any point in waiting for her to become symptomatic. The markers are telling us something. They’re telling us that our treatment is ineffective, so perhaps, at the best, if we don’t begin another treatment, maybe we should stop what we’re doing. On the other hand, then we’re simply waiting for her to become symptomatic. The nature of her disease is such that the imaging studies are not likely to be terribly helpful, and she was rather severely symptomatic in early 2003 with the first appearance of metastatic disease. Now, by analogy with other diseases, in which we don’t have a tumor size to measure — such as ovarian cancer — we do make our decisions on the basis of the marker. Testicular cancer would be a similar story. DR HUDIS: I don’t disagree. The differences here are subtle. You’re with the patient and you’re examining her and all your points are entirely valid. The dilemma is that soon we’ll be inflicting the toxicity of chemotherapy on a patient who no doubt will need it sooner or later, but maybe could have waited until later. DR LOVE: Before we move off the tumor markers, I’d like to get John’s take on them. Do you agree with Cliff’s point of view? DR MACKEY: I tend to be one of the unbelievers in tumor markers in breast cancer. At the end of the day, I think the clinical trials that guide our practice have almost universally been conducted without markers, which is why it’s such an open question. I’m not saying following markers is wrong. It’s just untested, so my preference is to wait for objective evidence of some type before switching off a potentially effective hormonal therapy. DR LOVE: Cliff, suppose two new lesions show up on a bone scan, with x-ray correlation. Now what? DR HUDIS: This person at some point needs chemotherapy. But I’m not even sure this is it. With a 15-year interval, or more, from when she was first diagnosed, the growth curve of her breast cancer is shallow enough that this is the kind of patient for whom I might reuse tamoxifen. I might try estrogens. I might try megestrol acetate. I might do everything I could when she’s asymptomatic to get through hormonal therapy. When the time came for chemotherapy, I would not follow the normal algorithm. This is the exact kind of patient for whom I would start with oral capecitabine because it would represent for her in many ways a less toxic alternative again (3.5). The therapy she gets first, statistically, is the therapy she’s likely to be on longest, and the toxicity profile for capecitabine — without alopecia or significant myelosuppression — would represent an advantage over other cytotoxic agents.
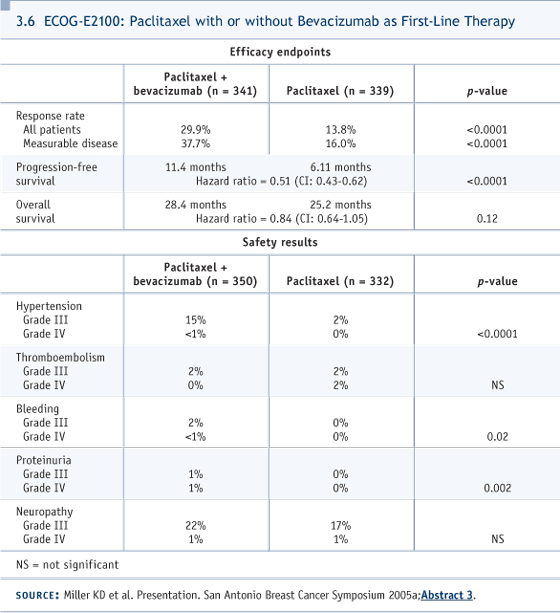
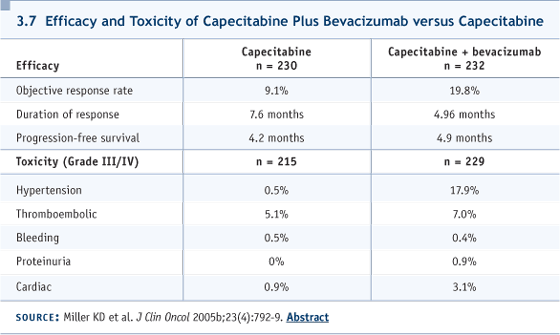
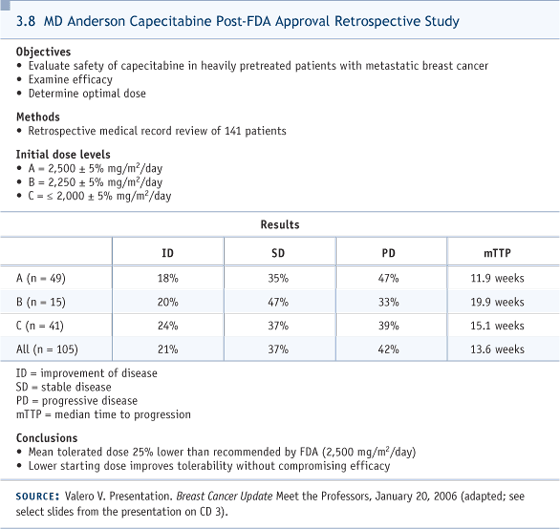
I would add that if somebody says, “I would use weekly paclitaxel” or “I would use docetaxel,” or “I would use anthracyclines or gemcitabine or vinorelbine,” no firm case can be made in any direction. I’m presenting a biased view based on my assessment of the toxicity profiles. DR LOVE: Cliff, putting aside reimbursement, would you use capecitabine alone or with bevacizumab? DR HUDIS: We don’t have data for first-line capecitabine with bevacizumab, but, in fact, when I give first-line chemotherapy in the Stage IV setting, I routinely try to use bevacizumab now, based on the ECOGE2100 trial (Miller 2005a, 2005b; [3.6]). They used paclitaxel. That’s probably a nonissue because bevacizumab, in general, is used with almost every chemotherapy agent across a variety of diseases. So I don’t have particular concerns about interactions, favorable or unfavorable. We have safety data for bevacizumab with capecitabine from Kathy Miller’s trial in the salvage setting, and despite the labeling of that trial as negative, it showed clear biologic signals of increased activity (3.7). So if the patient has no contraindication and is starting first-line chemotherapy and is HER2-normal, we routinely try to use bevacizumab. DR LOVE: What is the dose and schedule of capecitabine and bevacizumab in your regimen? DR HUDIS: We have two options. Usually we start with the standard schedule of three-week cycles, 14 days on. We use a lower-than-standard dose of capecitabine, aiming for 2,000 mg/m2 (3.8). We are studying a week-on, week-off schedule, which is used in other diseases and by clinicians anecdotally, but hasn’t been formally tested in breast cancer. We’ve been dose escalating, using flat dosing in a Phase I setting, and we’re going into Phase II shortly. DR LOVE: What about the dose and schedule of the bevacizumab? DR HUDIS: I would match it, for convenience, to the patient’s regimen. If she’s visiting every three weeks for initiation of capecitabine, then I administer 15 mg/kg of bevacizumab every third week, and so on.
DR LOVE: John, reimbursement, cost and regulatory issues aside, what do you think the best therapy would be for this woman in this situation? DR MACKEY: I would agree that recycling the hormones might be a very valid option for this lady. It’s been years since she’s been exposed to tamoxifen. Megestrol acetate is not a fun drug, but it could be considered as an option. If I were to use first-line chemotherapy, I would use capecitabine at 2,000 mg/m2, two weeks on, one week off. In Canada, we are not in a position, either from the approval standpoint or the financial standpoint, to administer bevacizumab for metastatic breast cancer in the absence of survival advantage. Our regulatory agencies require overall survival data before they’ll move on approval of expensive new therapies. DR LUNIN: I’d like to ask a question related to Dr Abel’s description of this case. For me, it just didn’t feel right. This woman burned through three hormonal therapies in a matter of two to three years after a 15-year disease-free interval. I got the sense that perhaps we weren’t measuring her response to those therapies as accurately as we should have. We were discarding therapies that were deemed ineffective, and maybe they weren’t effective, or perhaps something is different about the biology of her disease today than when she was first diagnosed. Has the tumor been rebiopsied?
DR ABEL: It was done on the specimen from the left axillary node. DR LUNIN: Okay, and is three months enough time to know if fulvestrant is working or not? Can you abandon fulvestrant after three months? DR HUDIS: These are complicated questions. First of all, if you go back to Dan Hayes, who did some of the initial work with CA 15-3, and you look at CEA, both as markers for progression, they do predict progression, but the problem is, the time to progression, clinically, can be out to seven months or longer from when the marker goes up. So it’s an early look at an inevitability. The challenge is to prove that early intervention in that setting makes a difference, and you have a lot of evidence that suggests it doesn’t. For example, early detection of metastatic disease in two randomized trials from the 1990s doesn’t lead to an improved outcome. By extension, early response to an inevitable future progression of disease may not improve outcomes, despite everybody’s hopes to the contrary. The second issue that I wanted to get to is duration of treatment. From bone-scan data, at least, we know that early observation of disease status after you begin a new hormone therapy can be misleading, for sure. Bone scans in particular can actually look worse, and we have reasonable evidence that blood test results can spike and fall in response to a new hormone therapy. Three months is a little long for that still to be true, but, again, in a patient with no symptoms, one would be inclined to hold tight and see what the shape of the curve is for the markers. Maybe they are going to spike and drop. DR ABEL: Is there a conflict between the concept of advocating late extended adjuvant therapy and not treating based on tumor markers. Is that conceptually coherent? DR HUDIS: I think so, because the patient population is quite different. In the extended adjuvant setting, overwhelmingly, you’re dealing with patients who are cured, so your toxicity profile matters a whole lot. In the metastatic setting, you’re palliating patients who will all die of their disease, and quality of life is their paramount issue. So I don’t think they’re the same at all.
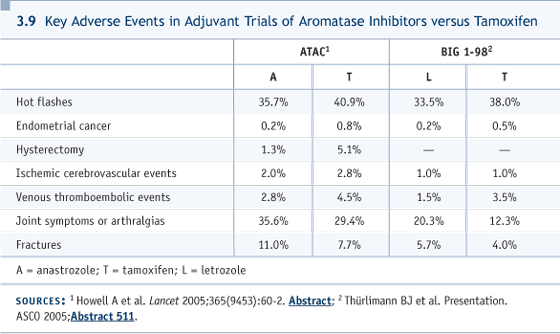
DR KUZUR: From my experience, most patients with bone-only disease when they become symptomatic will remain symptomatic, even with effective therapy. So I don’t see a big offense in following the markers as a guide for changing your therapy. I have a patient in a wheelchair, and I waited until she became symptomatic. She received radiation therapy and chemotherapy, and she’s still symptomatic. DR HUDIS: For every anecdote, an opposite anecdote exists. I’ve taken a patient out of a wheelchair with hormone therapy. Let me be very clear. I’m not advocating waiting until patients are symptomatic. I’m advocating waiting for objective progression of something other than the blood test. That’s the key difference. While in this case it worked nicely, if you’re using the blood test as a marker, have you objectively prolonged survival? That’s tough to prove. Have you improved her quality of life? That’s really tough to prove. Those are the real endpoints for Stage IV management. The world will change one day. We will have curative therapy, I believe, for Stage IV disease, and then jumping on this will matter more than it does right now. I want to give one more inch back, though. Without question, for disease that’s difficult to evaluate, sometimes the blood marker is all you have to go by. That’s true, but one should be very conservative about burning up a lot of therapies in asymptomatic patients. DR LOVE: John, if you treat this woman with capecitabine and she has a nice response for a year and then progresses, then what? DR MACKEY: Then we’re looking at second-line chemotherapy, and we have a number of relatively gentle options. In Canada, we like to use monotherapy if a person is asymptomatic or minimally symptomatic and has a relatively slow pace of disease. However, in this case you could also make an argument for combination therapy if she has had only one year of benefit from the taxane. That’s better than average, but at the end of the day, if she were highly symptomatic or had visceral disease at that point, we’d go into combinations. It’s not clear that there’s any particular advantage to sequencing the anthracyclines before or after the taxanes. She is anthracycline naïve and taxane naïve, so you have many options for her. The more gentle options, though, like vinorelbine, probably have lower response rates. Single-agent gemcitabine is very well tolerated. In some patients, even weekly epirubicin can be remarkably well tolerated for a long time. DR LOVE: If you’re using a taxane for first-line therapy — let’s say paclitaxel — putting aside the reimbursement issue, do you think bevacizumab should be added in? DR MACKEY: At present, with paclitaxel, in the best of all worlds, yes. I’d give weekly paclitaxel and bevacizumab. I think the data are quite strong, with the time to progression being very robust. It’s a little early to say whether overall survival will be shifted or not. I think, with further follow-up, we may see that. However, at present, the most recent analysis does not quite reach a statistically significant level of overall survival benefit, and it’s harder to leverage those drugs in the clinic. DR LOVE: Cliff, we’ll say that you used the capecitabine and bevacizumab and she had a two-year response, but then progressed. What do you do next? DR HUDIS: It’s the same issue. I weigh, at that time, her tumor burden and her symptoms against the toxicity profiles of the various drugs. If she still had an indolent disease pattern, I would go with one of the single-agent chemotherapy regimens, and I don’t have a strong bias for any one of them, frankly. We tend to use weekly paclitaxel, vinorelbine or gemcitabine, and the order is close to random. DR LOVE: What about keeping the bevacizumab going? DR HUDIS: I can’t do that right now. It goes a little bit to the biology. While debate continues endlessly about trastuzumab beyond progression of disease, that’s the sort of frame of reference we have. If a tumor is able to grow despite years of VEGF ligand blockade, one has to speculate that this tumor is growing independently of that treatment. It’s a treatment with some toxicities and expense and no evidence to suggest that you should use it beyond progression. DR LOVE: Dr Abel, your patient was treated before the bevacizumab data came out? DR ABEL: Yes. She received single-agent capecitabine at 2,000 mg/m2 and tolerated it well. She’s done well to the present time. Her tumor markers are down enough to maintain a level of comfort, and they’re staying down. DR LOVE: Cliff, if the patient stays like this for a while and then progresses, would you add bevacizumab on top of it? DR HUDIS: No, I would change the chemotherapy and add bevacizumab. DR DRAGON: Could you walk through the absolute benefit of using the aromatase inhibitor after five years for a population similar to Dr Abel’s patient — the T1B, and then the T1C population? DR LOVE: Rowan? DR CHLEBOWSKI: When you look at the numbers, it looks as if half the recurrences are yet to come. So the easiest way for me to figure it out now, while we’re waiting for data, is to apply the same argument you made when you decided to give the therapy initially, at least for the breast cancer risk reduction. Different interpretations about the toxicity profile may exist, but for the risk reduction, it seems that the same numbers should apply, but spread out over 10 years. DR LOVE: For practical purposes, Cliff, a woman comes in to you like this, who’s five years past her tamoxifen, and says, “What’s the chance over the next five years, I’m going to have a relapse?” DR HUDIS: I answer like Rowan does. I say, “It’s about the same as when you first came in.” DR LOVE: What’s the number with a T1B, one-centimeter tumor? DR HUDIS: For ER-positive disease of that size, node-negative, it’s probably in the six to 10 percent range over the next five years. DR LOVE: And if she had three positive nodes originally? DR HUDIS: The positive nodes make it easier on all counts, because in MA17, the planned analysis for node-positive disease shows a survival advantage (Goss 2003, 2005). DR LOVE: Okay. She has three positive nodes and is five years past her tamoxifen. She asks, “What’s the chance I’m going to relapse over the next five years?” DR HUDIS: I would have initially said that with a one-centimeter tumor and three involved lymph nodes, her five-year risk would have been up around 20 to 25 percent, and I would say that her risk in the next five years is probably pretty close to that. DR CHLEBOWSKI: I think this is where toxicity comes into play, because these are the numbers we’re going to be dealing with all the time now. In the old days, we saw patients with 10 positive nodes. In the Women’s Health Initiative, which does mammograms every year, we get 1.5-centimeter tumors. Instead of the expected 30 percent or so node-positive, we get 15 percent node-positive. That’s our reality for the future. If you’re a big believer in node-positive versus node-negative, you’re going to have no data set in the future, because the node-positive studies will have no patients. We’re going to be dealing with low-risk populations from now on, and I think the story is going to be the toxicity story, because efficacy’s pretty clear. DR LOVE: Rowan, can you briefly summarize the key differences in toxicity between tamoxifen and the aromatase inhibitors (3.9)? DR CHLEBOWSKI: The three life-threatening toxicities associated with tamoxifen — endometrial cancer, pulmonary emboli and stroke — are all reduced with anastrozole, but there are more joint symptoms and fractures. I’ll make the case that those symptoms may actually go away completely. Just as a reminder, none of the aromatase inhibitor trials had pretreatment screening with bone mineral density or ongoing protocol-defined intervention strategies. Eight percent of patients on the ATAC trial received bisphosphonates, almost all in the last couple of years, and almost all were in the United States. There are more data on fractures from Tony Howell in a letter to the Lancet. This is a preplanned analysis six months after stopping therapy, and the hazard ratio for hip fractures is the same between anastrozole and tamoxifen. Is that biologically plausible? We know one of the problems of estrogen plus progesterone, or estrogen use for bone mineral density maintenance, is that once you stop, you get accelerated bone loss at two to three times the age-adjusted rate. Could we be seeing accelerated bone recovery after we release the estrogen suppression from anastrozole? They’re going to have some tests in bone mineral density that’ll help answer that question. Also, the hip fractures, after 68 months, are the same: one percent for tamoxifen and one percent for anastrozole, even without intervention. If, with modern management strategies for bone health, we’ll be able to prevent most of the factures, then the current balance for side effects is overwhelmingly in favor of the aromatase inhibitors. DR LOVE: What do we know about the ability of bisphosphonates to prevent the bone loss that occurs with aromatase inhibitors?
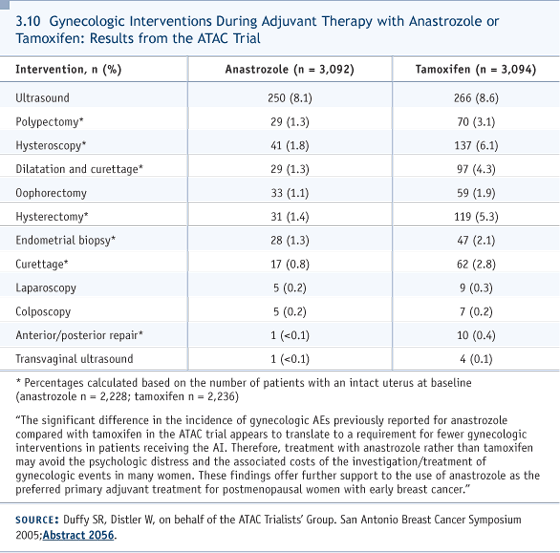
DR CHLEBOWSKI: There are a couple of ways of showing that bisphosphonates work. ABCSG Trial 12, interestingly, took premenopausal women with estrogen receptor-positive disease and made them postmenopausal with goserelin, and then administered anastrozole versus tamoxifen. So you’re making women postmenopausal very suddenly, and what happens when you give them either zoledronic acid or not? If you don’t give them zoledronic acid, tamoxifen isn’t able to prevent, fully, this rapid loss of bone that occurs, and there was more bone loss with anastrozole. However, bone loss was completely abrogated by zoledronic acid four milligrams every six months. The Z-FAST trial had about the same results. I’m involved with helping write up the ASCO bone health section that was published in 2003. We’re in the process of updating it now, saying everybody gets a bone mineral density. You have the option to follow or treat patients with osteopenia, treat women with osteoporosis and repeat bone mineral density annually. We believe that only about 20 percent of women then would need a bisphosphonate on aromatase inhibitors, but we’ll have to see if that’s the case or not. DR LOVE: One of the interesting findings that came out of the most recent update of the ATAC trial at 68 months was a marked increase in the rate of hysterectomy in women who received tamoxifen compared to those who received anastrozole. Can you talk about the magnitude of this increase and why you think it occurred? DR CHLEBOWSKI: The numbers were somewhat over five percent of women on tamoxifen having hysterectomies and 1.4 percent on anastrozole. The telling additional observation, which was presented at San Antonio (3.10) just this year, was that the number of women having gynecological interventions — mostly surgical biopsy procedures of one kind or another, commonly endometrial aspiration biopsies — was about 600 in women on tamoxifen versus a hundred or so women on aromatase inhibitors. Six hundred out of 3,000 is a large number. I believe women on tamoxifen will get some vaginal discharge and have some symptoms that lead to a gynecologic workup. A gynecologic workup can sometimes identify problems — not only endometrial cancer and fibroids — that will lead to these interventions. That is a recently emerging, additional side effect that more women on tamoxifen experience compared to women on aromatase inhibitors.
|
Case 1: from the practice of Leon H Dragon, MD Case 2: from the practice of Sheryl R Simon, MD Case 3: from the
practice of Howard R Abel, MD Case 4: from the practice of Mary
Ann K Allison, MD Case 5: from the practice of Richard S
Zelkowitz, MD
|
| Terms of Use and General Disclaimer Copyright © 2006 Research To Practice. All Rights Reserved |